Tuberculosis (TB) is one of the world’s
leading causes of death from a single infectious agent, second to human immunodeficiency
virus (HIV). However, TB-HIV co-infection is a serious problem because current
TB drugs (such as rifampin) induce hepatic metabolic enzymes (such as CYP2C9
and CYP3A4) that degrade anti-retroviral drugs, which in turn renders patients
in an immunocompromised state that accelerates the progression of their TB.

In their search for new anti-TB drug leads
that do not induce CYP activation, the Looper group and collaborators identified
the known natural product amicetin (Figure 1) and found that it
possesses potent anti-TB activity (IC50 = 0.24 µm), but less than
3-fold induction of CYP3A4 and CYP2D6 (rifampin induction levels are 47- and
6-fold, respectively). Therefore, the group decided to utilize to modify the
pharmacophore of this natural product to address the need for new TB therapies.
Although amicetin and its natural relatives have been shown to inhibit protein
synthesis by binding to the prokaryotic ribosomal P-site, the molecular nature
of these interactions have not been fully elucidated. The group co-crystallized
purified amicetin with the 70S subunit of the ribosome of Thermus
thermophilus (Tth) and generated a crystal structure at 3.5 A
resolution (Figure 2).

On the basis of these findings, the
authors were able to hypothesize that the acid labile cytosine glycosidic
linkage could be removed, since its hydrolysis products cytimidine and cytosamine
are both inactive, and the disaccharide moiety could be simplified such that
the synthetic chemistry is amenable to basic medicinal chemistry tactics. Their
synthetic approach is summarized in Scheme 1. Notably, the removal of
the disaccharide significantly simplifies the synthetic route, and the only
chiral information in the synthetic analogues is embedded in commercially
available α-methylserine.

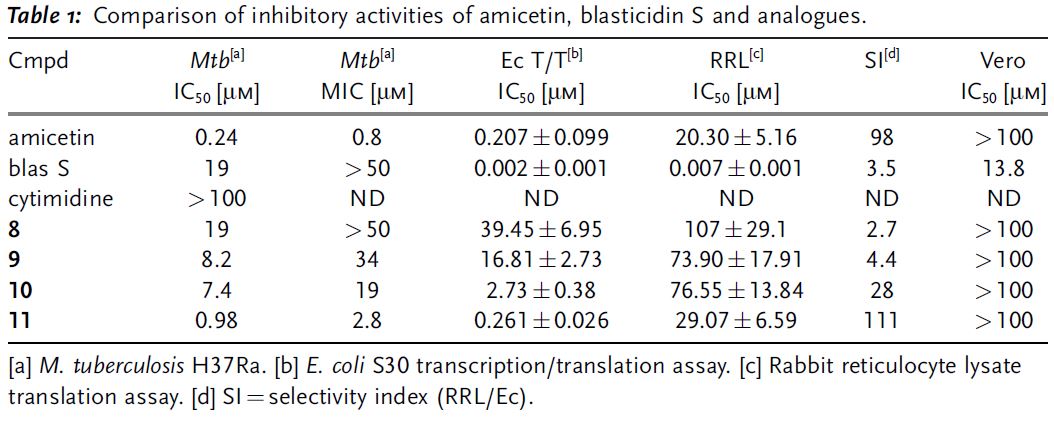
Remarkably, compound 11 is only ~4
fold less active than amicetin, despite the removal of 8 stereogenic centers.
This analogue also maintained selectivity for inhibiting bacterial (vs.
eukaryotic) translation, in contrast to blasticidin
S (Table 1). Remarkably, no cross-resistance was
observed, and 11 maintains activity
against TB strains resistant to rifampicin (RIFR), isoniazid (INHR),
and levofloxacin (FQR), as shown in Table 2. The activity of
these analogues is limited to TB and some gram positive bacterial strains,
although the group is currently working on extending the spectrum of activity
of these compounds.

This study
provides the first structural characterization of ribosomal P-site inhibitor
that selectively inhibits prokaryotic translation. In addition, the study demonstrated
that structurally simplified analogues can provide tractable leads for new anti-TB
therapies.
Reference:
Serrano, C. M.;
Kanna-Reddy, H. R.; Eiler, D.; Koch, M.; Tresco, B. I. C.; Barrows, L. R.;
VanderLinden, R. T.; Testa, C. A.; Sebahar, P. R.; Looper, R. E. Angew. Chem.
Int. Ed., 2020 (Just Accepted) doi: 10.1002/anie.202003094.
Comments
Post a Comment